Deriving PSI3/5 values from splicing scores#
This notebook demonstrates how to derive Percentage Spliced In (PSI) values from predicted splice junction counts.
PSI_5 typically refers to the usage of a 5’ splice site (donor) relative to alternative donors, while PSI_3 refers to the usage of a 3’ splice site (acceptor).
Imports#
# @markdown Run this cell to install AlphaGenome.
from IPython.display import clear_output
! pip install alphagenome
clear_output()
from alphagenome import colab_utils
from alphagenome.data import gene_annotation
from alphagenome.data import genome
from alphagenome.data import transcript as transcript_utils
from alphagenome.models import dna_client
from alphagenome.models import variant_scorers
from alphagenome.visualization import plot_components
import matplotlib.pyplot as plt
import pandas as pd
Load the model#
dna_model = dna_client.create(colab_utils.get_api_key())
Score a variant#
Score a variant near a known splice site:
# Define a variant near a splice site in the BRCA2 gene.
variant = genome.Variant(
chromosome='chr22',
position=36201698,
reference_bases='A',
alternate_bases='C',
)
# Create a 1MB interval centered on the variant.
interval = variant.reference_interval.resize(dna_client.SEQUENCE_LENGTH_1MB)
# Predict junction counts for both REF and ALT alleles
output = dna_model.predict_variant(
interval=interval,
variant=variant,
requested_outputs=[dna_client.OutputType.SPLICE_JUNCTIONS],
ontology_terms=['CL:0000084'],
)
There are 107 unique junctions predicted across 2 tracks in this tissue:
print(f"Junctions shape: {output.reference.splice_junctions.junctions.shape}")
print(f"Value shape: {output.reference.splice_junctions.values.shape}")
Junctions shape: (107,)
Value shape: (107, 2)
There are two tracks because splice junction counts are predicted from RNA-seq data, and there are two types of RNA-seq data available for ‘CL:0000084’:
polyA RNA-seq for mature mRNA splicing because it enriches for fully processed transcripts.
Total RNA-seq captures pre-mRNA and nascent transcripts, which can sometimes look “messier” because splicing is still in progress.
output.reference.splice_junctions.metadata
| name | ontology_curie | biosample_name | biosample_type | biosample_life_stage | gtex_tissue | data_source | Assay title | |
|---|---|---|---|---|---|---|---|---|
| 0 | junction_CL:0000084 polyA plus RNA-seq | CL:0000084 | T-cell | primary_cell | adult | encode | polyA plus RNA-seq | |
| 1 | junction_CL:0000084 total RNA-seq | CL:0000084 | T-cell | primary_cell | adult | encode | total RNA-seq |
Derive the PSI values#
Create a function to calculate PSI values from splice junction counts for either RNA track:
def compute_psi_from_raw(junction_data, track_idx):
# Create the dataframe from known attributes
data = []
for i, j in enumerate(junction_data.junctions):
# Determine Donor and Acceptor based on strand
if j.strand == '+':
donor, acceptor = j.start, j.end
else:
donor, acceptor = j.end, j.start
data.append({
'donor': donor,
'acceptor': acceptor,
'count': junction_data.values[i, track_idx],
})
df = pd.DataFrame(data)
# Pivot into the matrix: Rows = Donors, Cols = Acceptors
matrix = df.pivot_table(
index='donor', columns='acceptor', values='count', fill_value=0
)
# PSI5: Normalize row-wise (Percent of donor usage for each acceptor)
# PSI5 = Count / (Total counts for that Donor)
psi5 = matrix.div(matrix.sum(axis=1), axis=0).fillna(0)
# PSI3: Normalize column-wise (Percent of acceptor usage for each donor)
# PSI3 = Count / (Total counts for that Acceptor)
psi3 = matrix.div(matrix.sum(axis=0), axis=1).fillna(0)
return psi5, psi3
Select RNA track and calculate:
# We can use either RNA track for PSI values
titles = (
output.reference.splice_junctions.metadata['Assay title']
.astype(str)
.tolist()
)
polya_idx = next(
(i for i, t in enumerate(titles) if 'polya' in t.lower()), None
)
total_idx = next(
(i for i, t in enumerate(titles) if 'total' in t.lower()), None
)
# Indecies
print(f'Indices found -> PolyA: {polya_idx}, Total: {total_idx}')
# Calululate PSI
psi5_total, psi3_total = compute_psi_from_raw(
output.reference.splice_junctions, polya_idx
)
Inspect results#
print(
'The proportion of total splicing activity at a specific acceptor site that'
' is contributed by a given donor'
)
display(psi3_total.head())
print(
'The proportion of total splicing activity at a specific donor site that is'
' directed to a given acceptor'
)
display(psi5_total.head())
The proportion of total splicing activity at a specific acceptor site that is contributed by a given donor
The proportion of total splicing activity at a specific donor site that is directed to a given acceptor
| acceptor | 36191912 | 36195432 | 36195437 | 36197831 | 36198896 | 36199369 | 36199376 | 36199608 | 36200291 | 36201796 | 36201851 | 36201888 | 36202055 | 36202094 | 36202933 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| donor | |||||||||||||||
| 36192925 | 0.018779 | 0.000000 | 0.000000 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| 36195094 | 0.016944 | 0.000000 | 0.000000 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| 36195310 | 0.908919 | 0.000000 | 0.000000 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| 36195912 | 0.004811 | 0.093950 | 0.016829 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| 36197944 | 0.005078 | 0.087348 | 0.017657 | 0.100797 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| acceptor | 36191912 | 36195432 | 36195437 | 36197831 | 36198896 | 36199369 | 36199376 | 36199608 | 36200291 | 36201796 | 36201851 | 36201888 | 36202055 | 36202094 | 36202933 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| donor | |||||||||||||||
| 36192925 | 1.000000 | 0.000000 | 0.000000 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| 36195094 | 1.000000 | 0.000000 | 0.000000 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| 36195310 | 1.000000 | 0.000000 | 0.000000 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| 36195912 | 0.153461 | 0.215491 | 0.631047 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| 36197944 | 0.129979 | 0.160791 | 0.531372 | 0.177858 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
Visualise Sashimi plot#
As we are considering splicing, we may want to plot more than just protein coding and MANE transcripts. Choose a relevant extractor, but be aware that including all possible transcripts can result in a crowded annotation:
# The GTF file contains information on the location of all transcripts.
# Note that we use genome assembly hg38 for human.
gtf = pd.read_feather(
'https://storage.googleapis.com/alphagenome/reference/gencode/'
'hg38/gencode.v46.annotation.gtf.gz.feather'
)
# All transcripts
transcript_extractor_all = transcript_utils.TranscriptExtractor(gtf)
# All protein-coding transcripts
gtf_protein = gene_annotation.filter_protein_coding(gtf)
transcript_extractor_protein = transcript_utils.TranscriptExtractor(gtf_protein)
# All MANE transcripts
gtf_mane = gene_annotation.filter_to_mane_select_transcript(gtf)
transcript_extractor_mane = transcript_utils.TranscriptExtractor(gtf_mane)
# All MANE protein-coding transcripts
gtf_mane_protein = gene_annotation.filter_protein_coding(gtf)
gtf_mane_protein = gene_annotation.filter_to_mane_select_transcript(
gtf_mane_protein
)
transcript_extractor_mane_protein = transcript_utils.TranscriptExtractor(
gtf_mane_protein
)
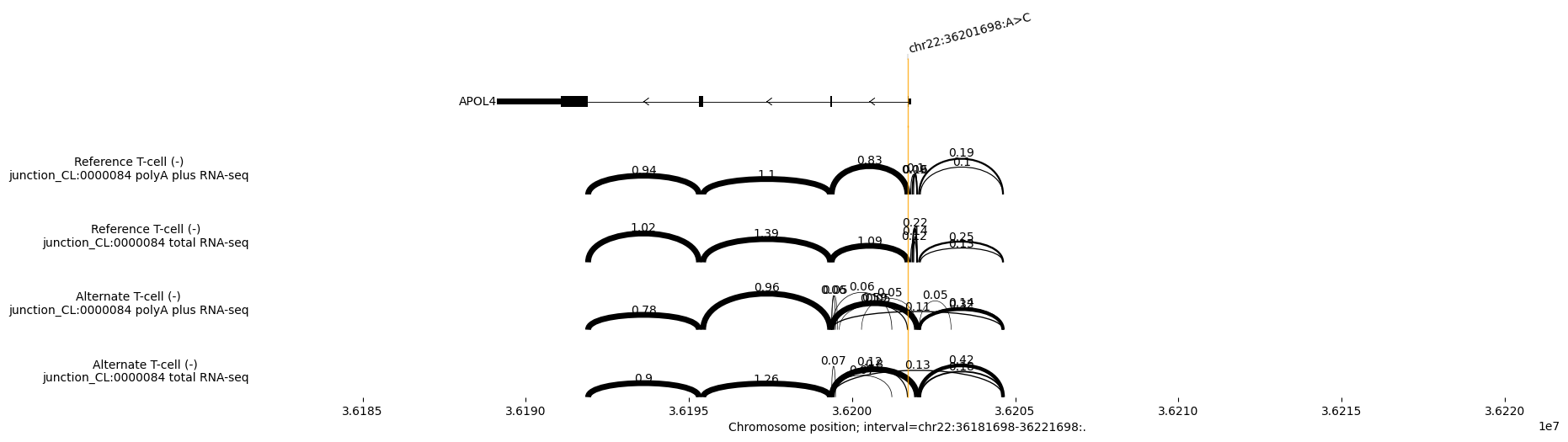
Build the Sashimi plot. The numbers above the junctions in the Sashimi plot are the predicted normalised read counts per donor/acceptor pair (quantitative signal strength). These are NOT the PSI values (proportions):
# We will plot for both polyA and total RNA-seq
ref_output = output.reference
alt_output = output.alternate
# Plot
_ = plot_components.plot(
[
# Transcript annotations
plot_components.TranscriptAnnotation(
transcript_extractor_mane.extract(interval)
),
# Sashimi ref and alt plots
plot_components.Sashimi(
ref_output.splice_junctions,
ylabel_template='Reference {biosample_name} ({strand})\n{name}',
color='skyblue',
),
plot_components.Sashimi(
alt_output.splice_junctions,
ylabel_template='Alternate {biosample_name} ({strand})\n{name}',
color='red',
),
],
# Annotate the variant position as a vertical line across all panels
annotations=[plot_components.VariantAnnotation([variant])],
interval=interval.resize(40000),
)
plt.show()